Help
Web server usage
The user can query protein alternative splice-forms derived from a given gene or transcript ID based on similarity from precalculated datasets by clicking on the “Explore our results” button.
Alternatively, it is possible to upload a DE transcript dataset of choice by clicking on the “Upload your results” button which will redirect you to an upload window.

The uploaded DE transcript dataset should contain information about transcripts in each row; the first column (transcript) will contain the ENST IDs, followed by the column ‘gene_symbol’ and then with two columns containing PPDE and log-fold changes of those transcripts in each tissue or condition. It is possible to have multiple such conditions/tissues by adding more columns. Please click here for an example input file.
The search bar suggests all the canonical isoform available for a given gene symbol.
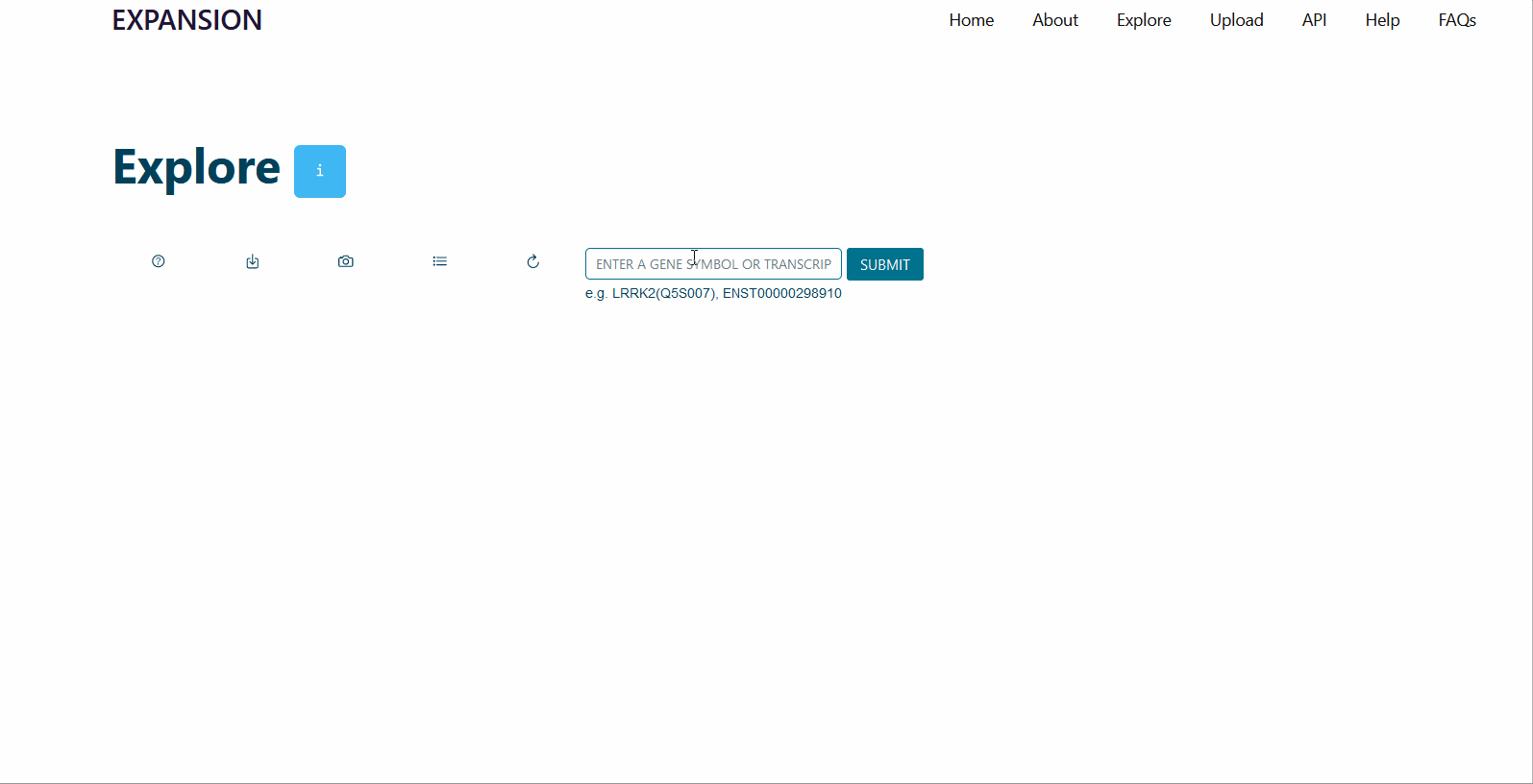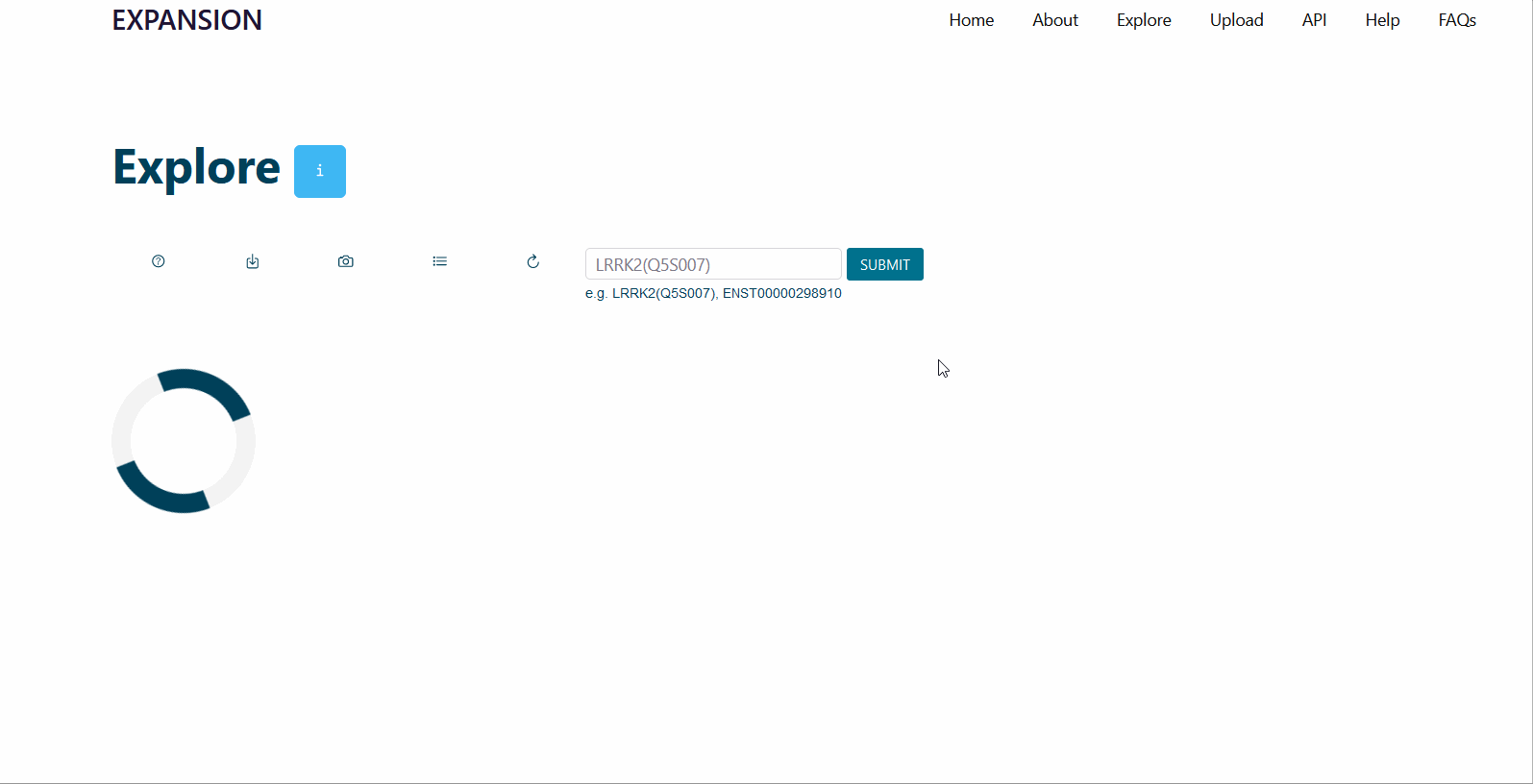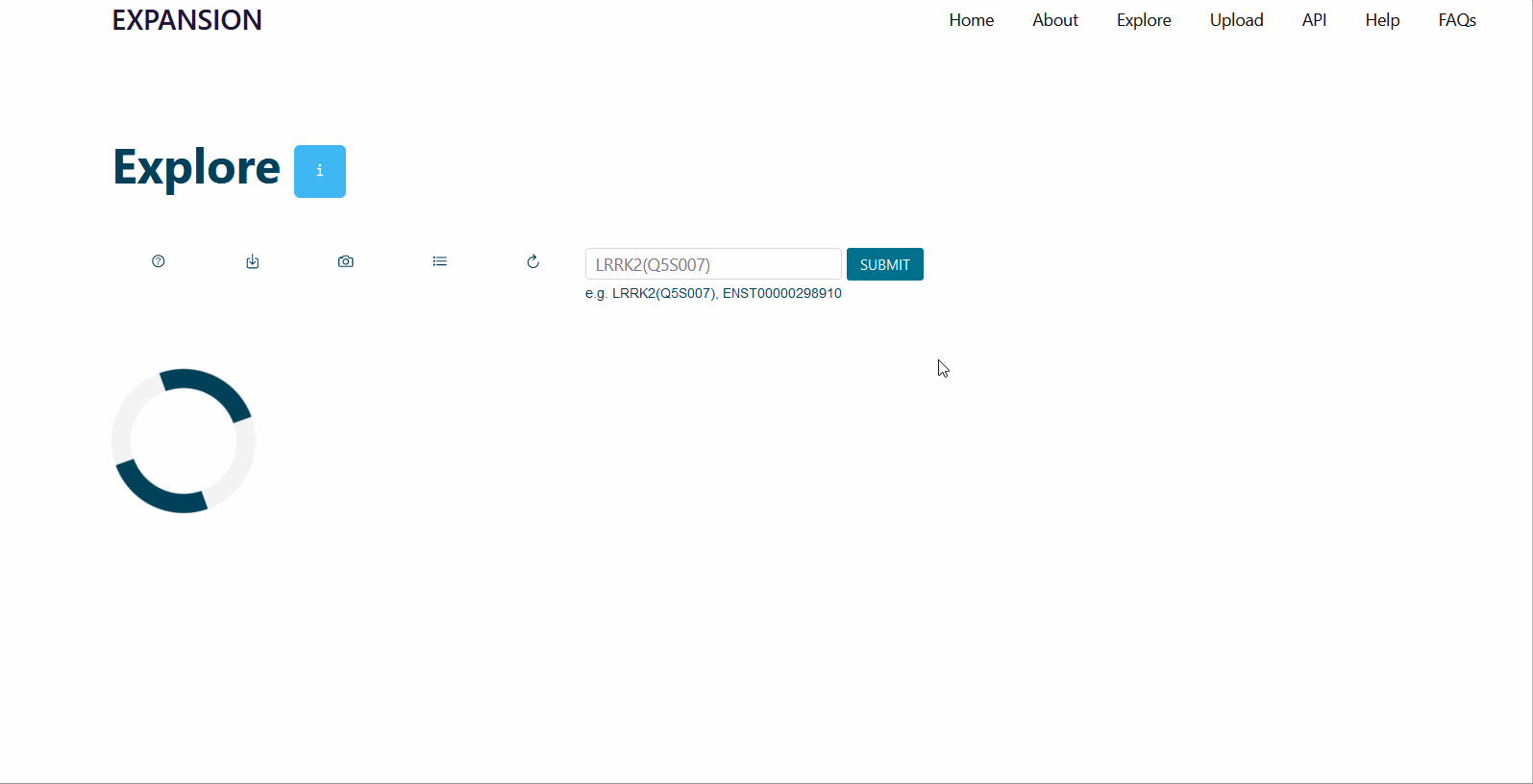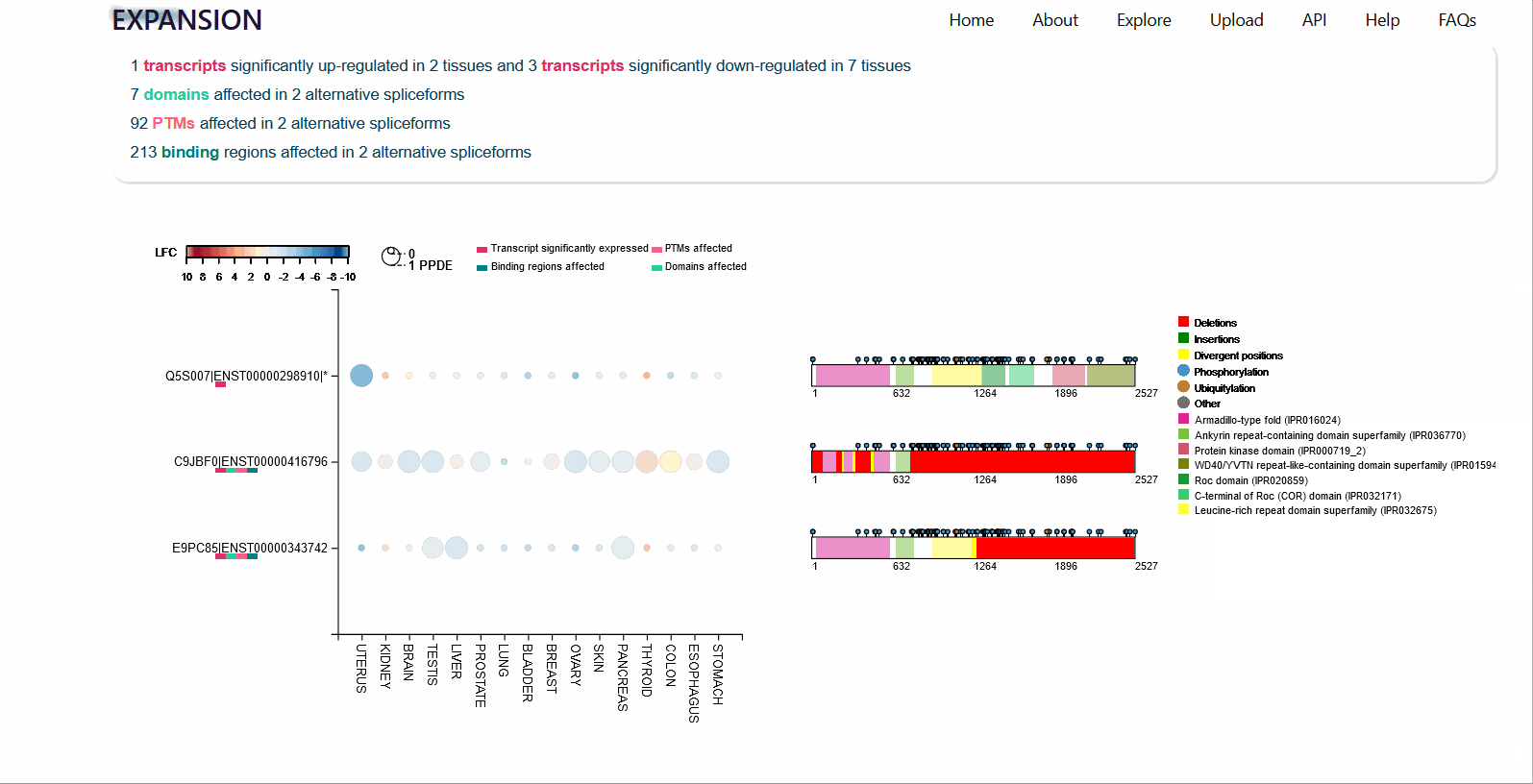
Differential expression of transcripts (rows) in each cancer tissue (columns), where each circle is colored based on log-fold change (TCGA over GTEx) while diameter is proportional to significance. Scrolling over each circle provides detailed information about DE values. By clicking on the transcripts (rows), you will be redirected to its UniProt page.
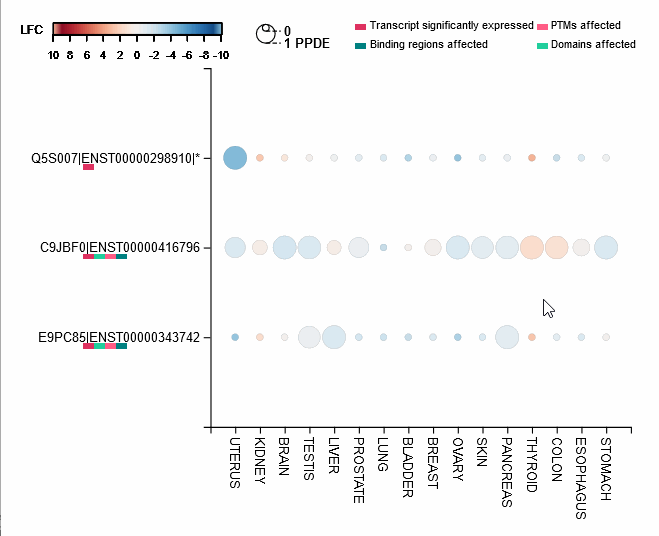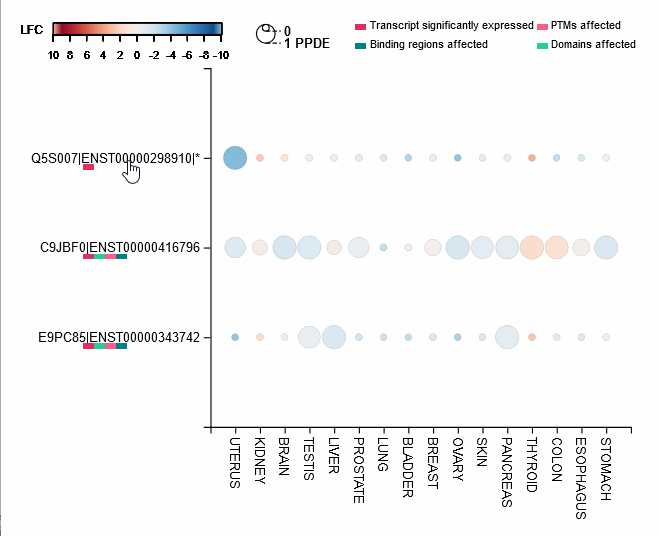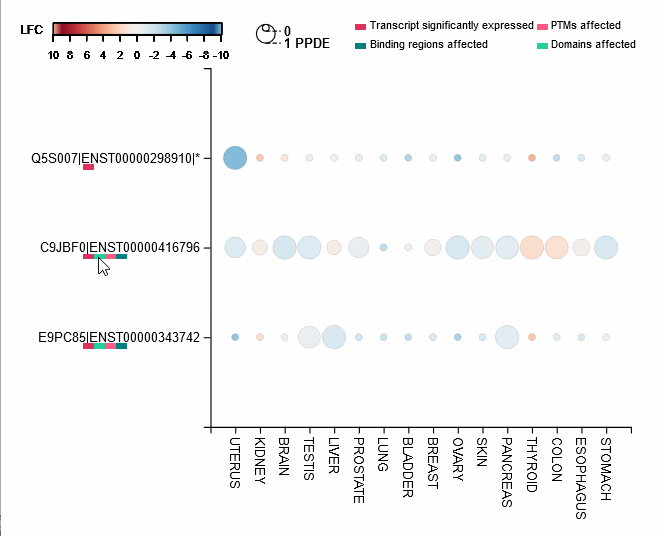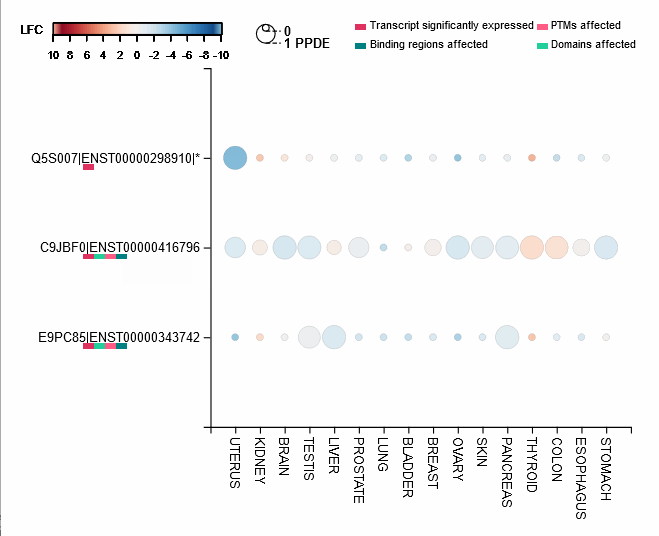
For each protein-coding transcript in the DE dataset, cartoon panels on the right provide information about splicing variation (i.e. insertion, deletion or divergence) affecting domain architecture as well as PTMs, respectively represented through colored boxes and lollipops on the canonical protein sequence. It is possible to visualize more information about each domain or PTM by scrolling over their region. The export button allows to download both the bubble plot as well as the domain architecture diagram in SVG format and PNG format.
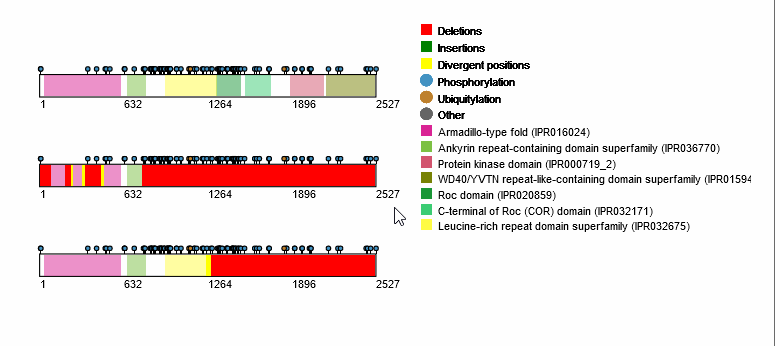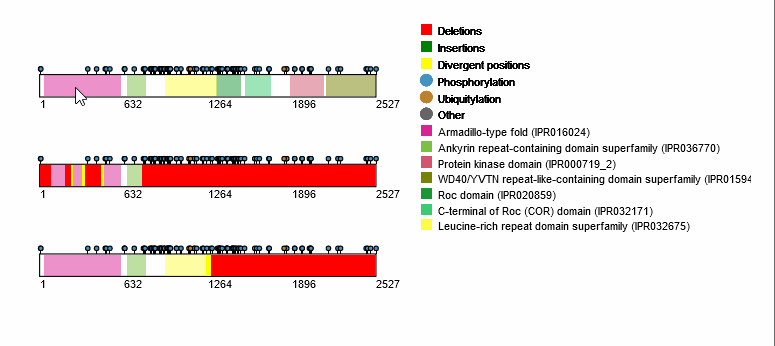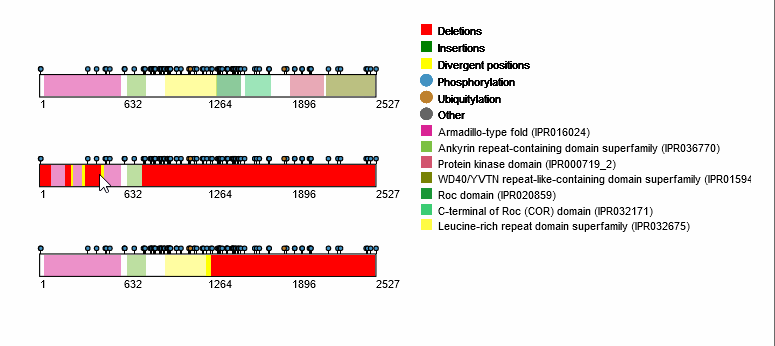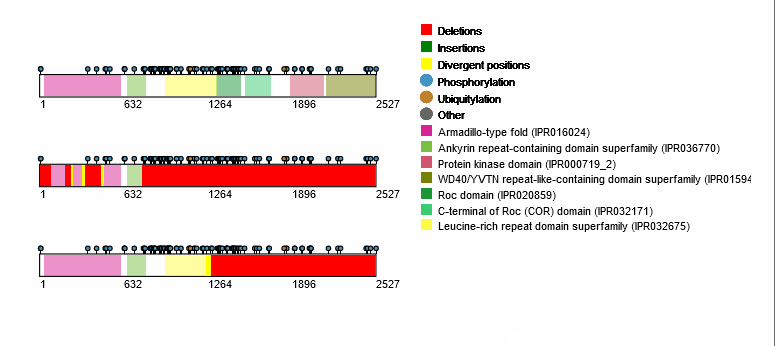
An MSA viewer will appear when you click on the domains, PTMs, or divergences. By clicking on the transcripts, you will be redirected to its UniProt page.

A central panel provides information about interaction networks (IntAct) mediated by the protein isoform considered. The number of interactors can be tuned using toggles for MI score (default MI score=0.2), interaction type (i.e. direct, physical or association) and cutoff for maximum number of isoform specific interactors (default=50). Each node represents a protein and can be expanded to visualize its domain architecture. If the binding region information is provided by IntAct, region-specific interaction edges are drawn on the network. If a splicing event affects a binding region, the corresponding edge in the network is highlighted in red to ease the interpretation of splicing functional consequence.
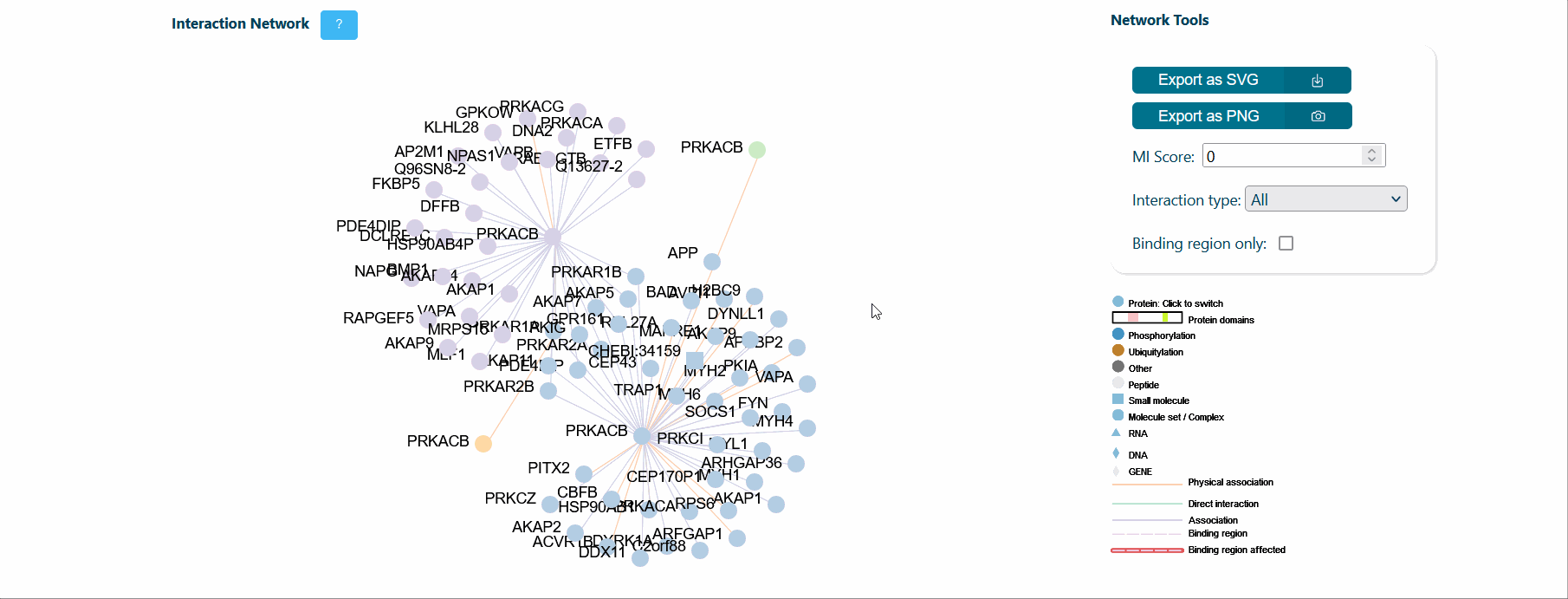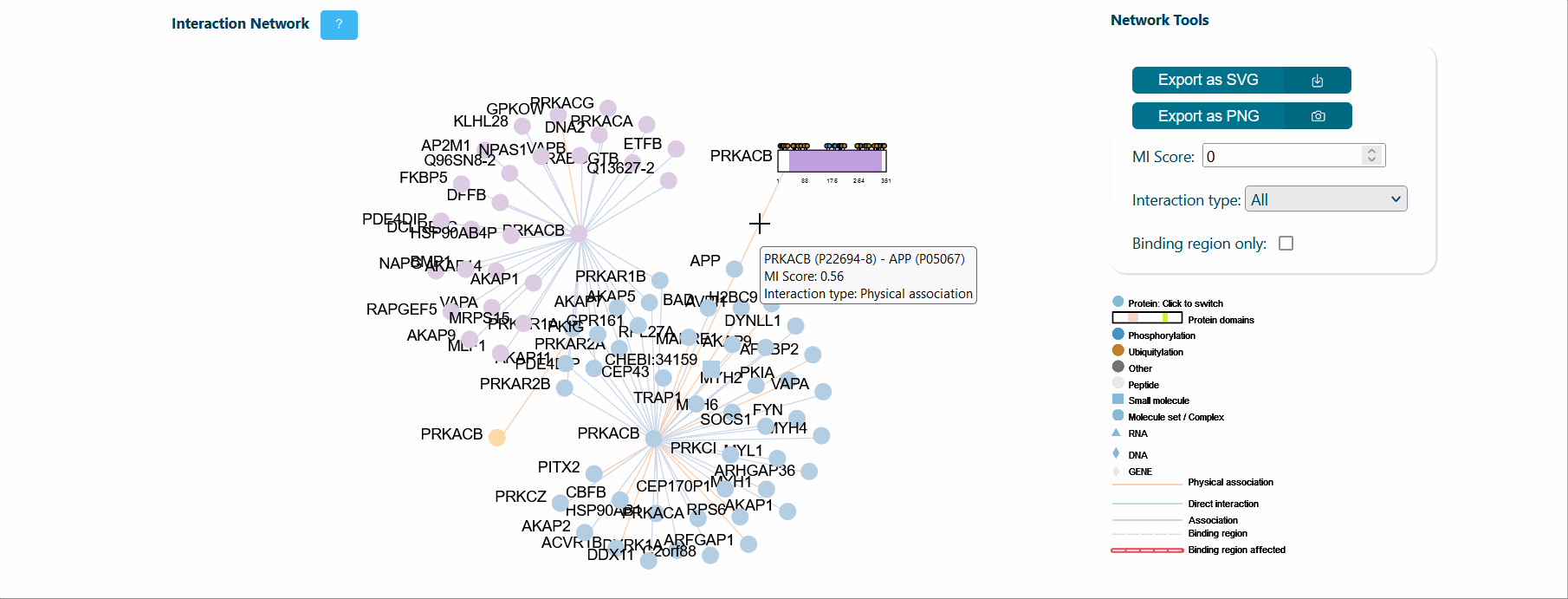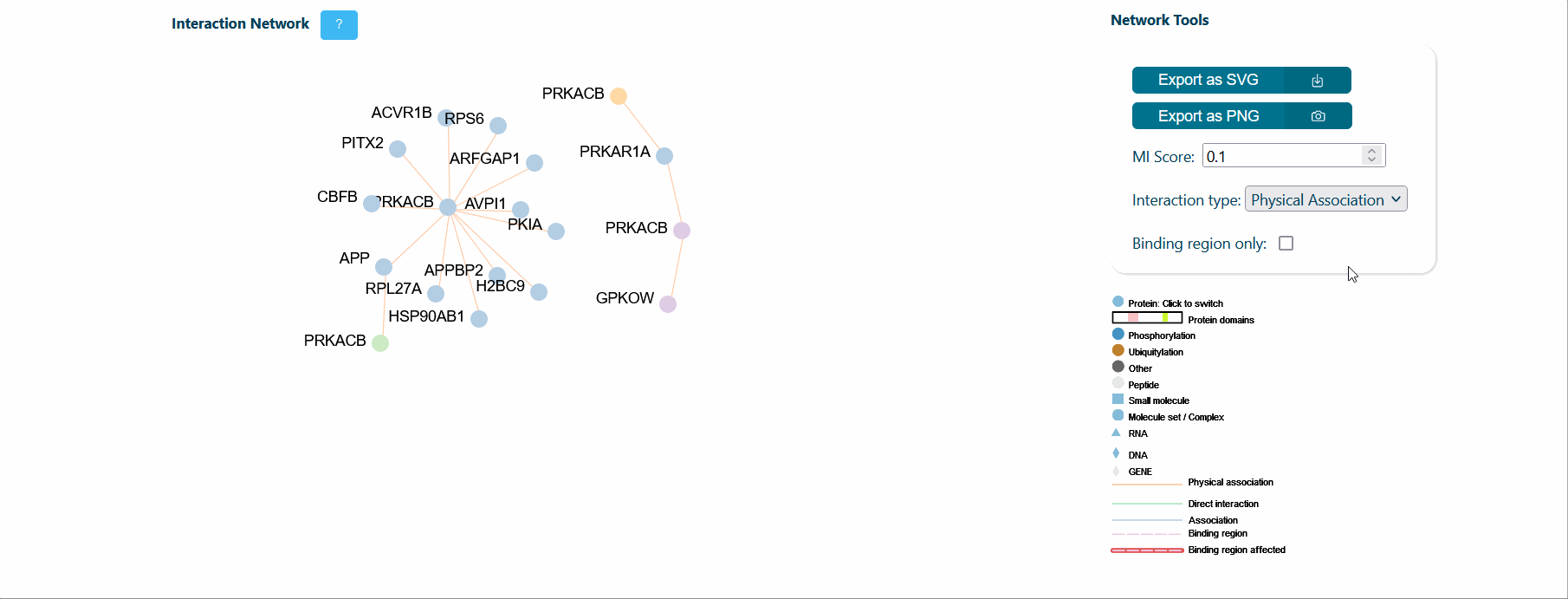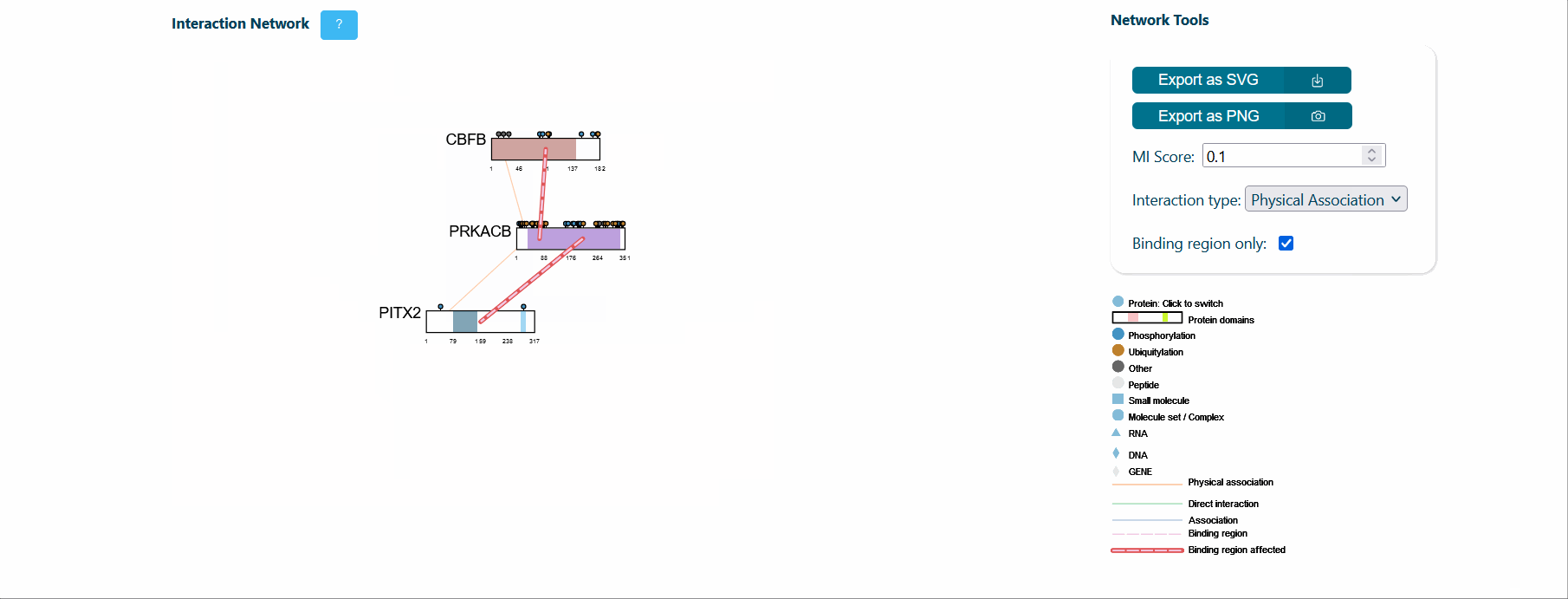
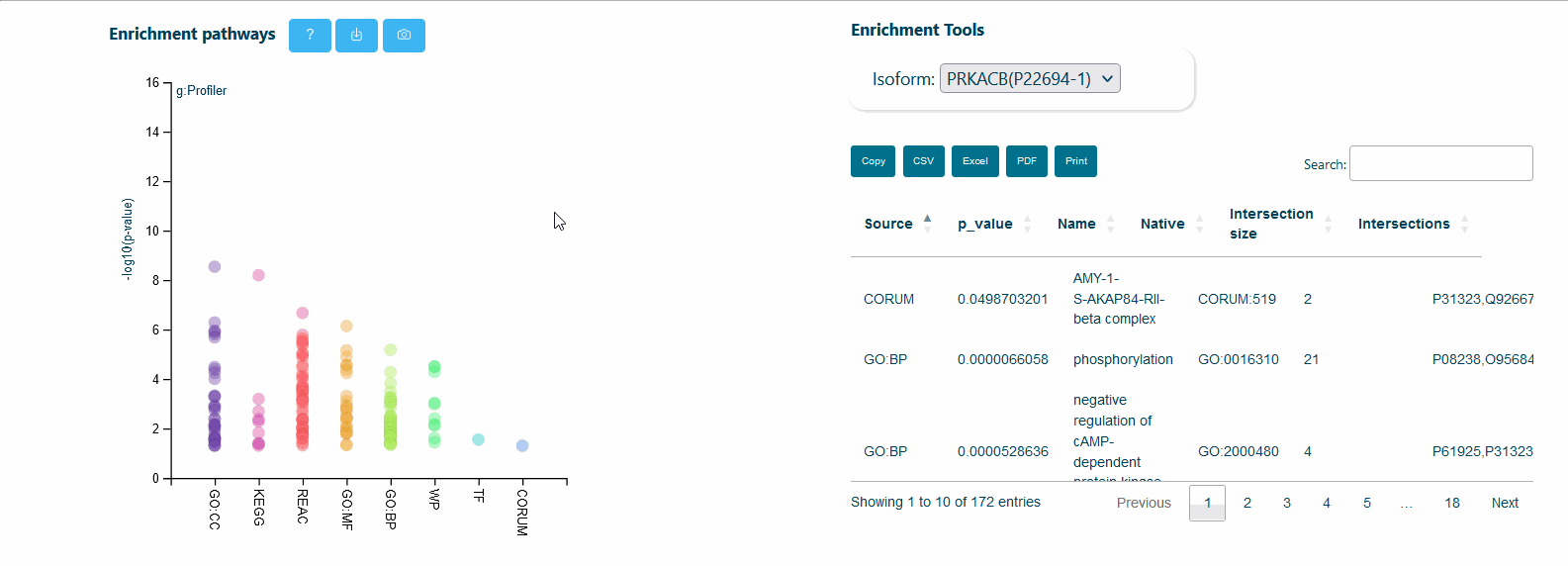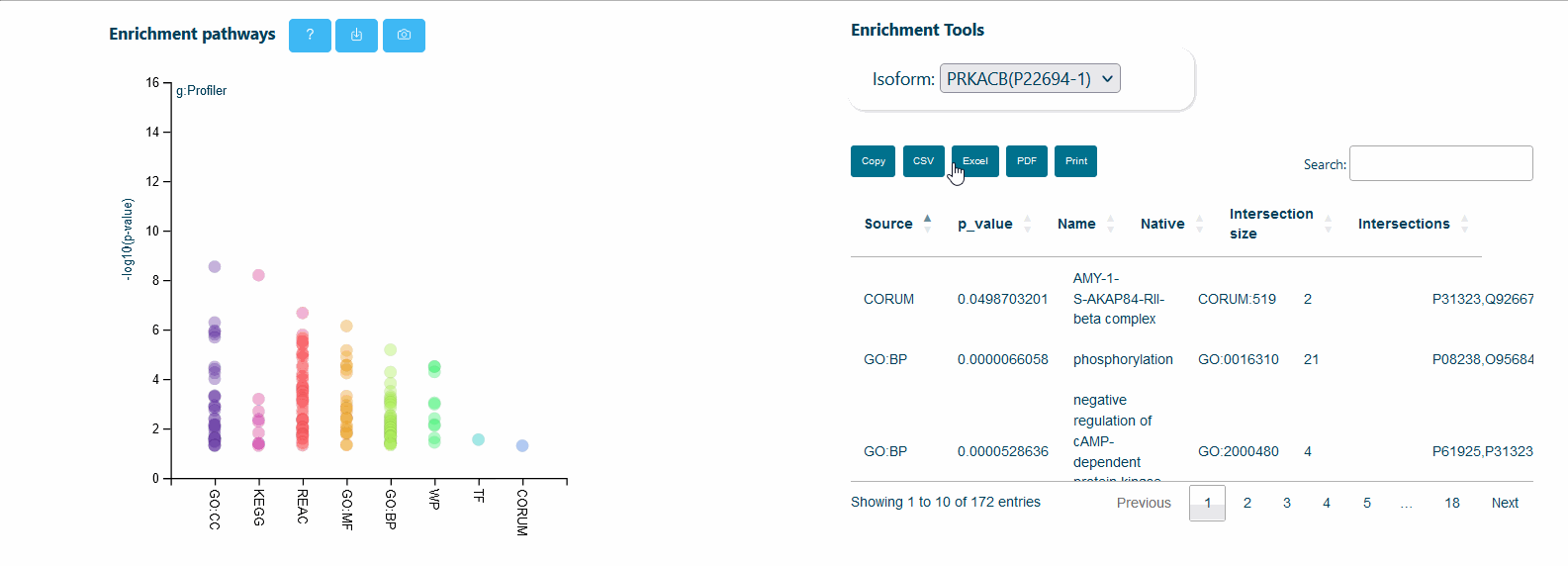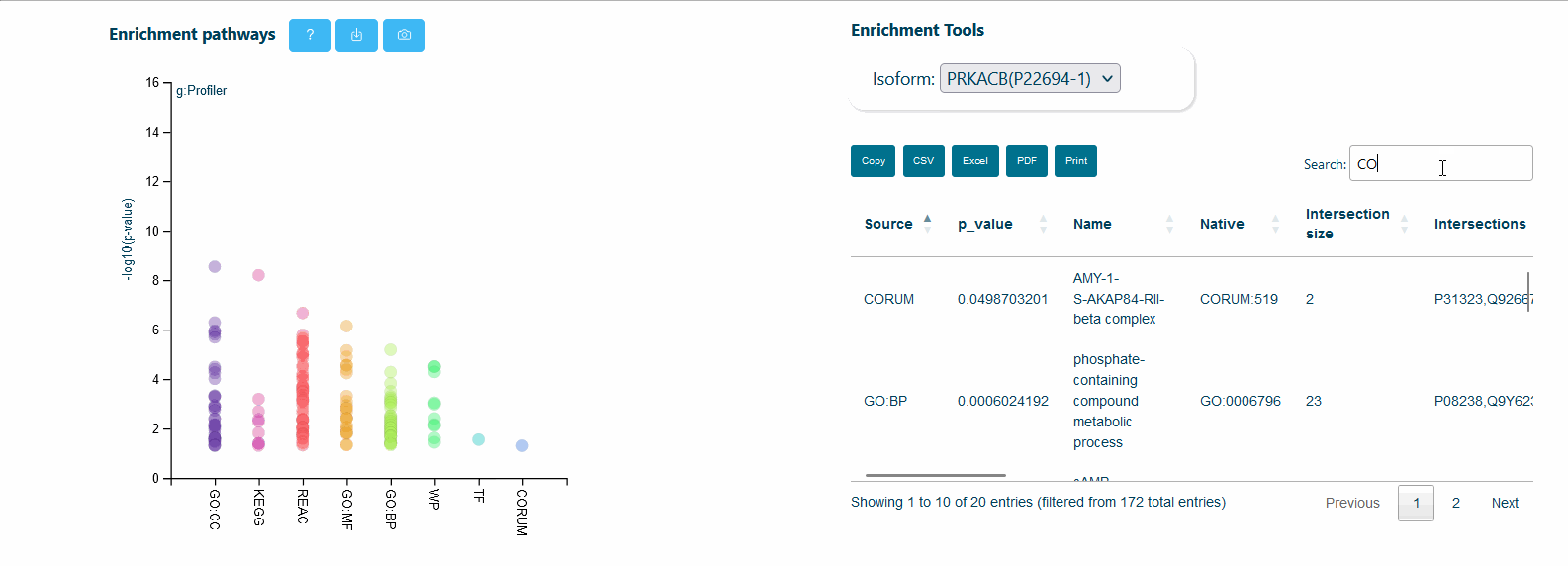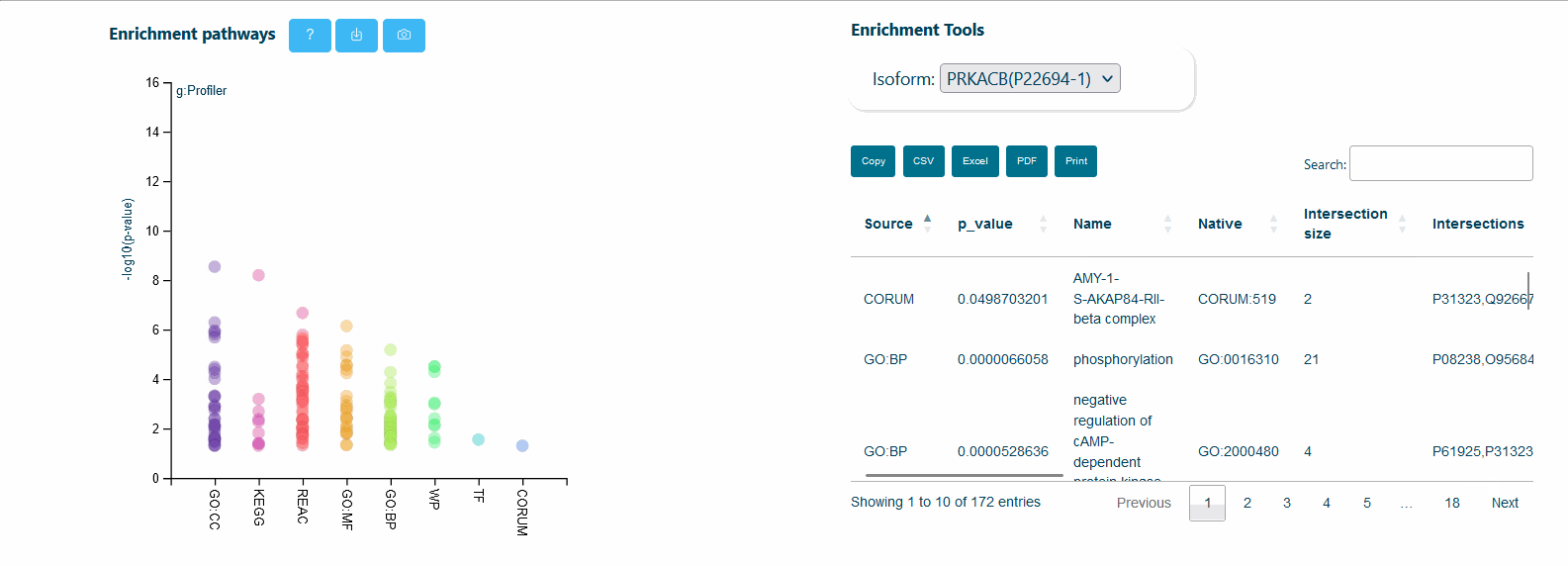
It is also possible to visualize over-representation analysis (ORA) of functional categories, computed via the g:profiler python library (https://pypi.org/project/gprofiler-official/), of the genes in the network. Whenever isoform-specific interactors are present in the network, ORA can be calculated for each isoform-specific group, allowing the comparison of distinct biological processes mediated by isoform-specific interactors.
API
The EXPANSION API — available at https://api.expansion.bioinfolab.sns.it — is our API that allows for programmatic access to our data.
You can query:
- /de: Differentially expressed (DE) protein-coding transcripts from cancer genomics
- /da: Provides information about splicing variation (i.e. insertion, deletion or divergence) affecting domain architecture as well as PTMs
- /ppi: PPI Interaction networks (IntAct) mediated by the protein isoform
- /ora: Over-representation analysis (ORA) of isoform-specific interactors categories computed via the g:profile
- /all: Provides a JSON of all analysis available in our web server for given a gene
For more information, please refer to the API page.